手动从GEO下载预处理Matrix文件并进行简单分析
编者:龙飞、汪宣佚、柴子轩;补充校对:liumwei
1. 首先搭建R环境
Windows下的R环境,请参考这个页面进行。若想重现本文相关操作和信息输出结果,请参看该页面进行R版本安装与配置进行。同时,本文所涉及的GEOquery和limma包安装,均借助Bioconductor下的BiocManager来进行,因此,请务必确认您已进行了如下操作:
>if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
如果上述执行有错误,请回到这个页面查看解决办法。
2. limma和ggplot2包的安装
在RStudio Console界面的>提示符后输入如下命令,并回车执行:
>BiocManager::install("limma")
>BiocManager::install("ggplot2")
提示:命令输入后,记得回车执行;中文输入法状态下,可能会出现不能复制粘贴命令(最好采用shift+ctrl+v粘贴),或手动不能输入命令。此时切换英文输入法状态即可。如果出现包下载速度很慢的状态,请回到这个页面查看解决办法。同时,安装ggplot2,可能会出现“Installation path not writeable, unable to update packages: survival”的错误,此时需要使用管理员权限打开RStudio,然后安装survival:
>BiocManager::install()
>BiocManager::install('survival')
如果安装limma也出现类似的错误,解决方式同上。
3. 加载相应的R包
>library('Biobase')
>library('limma')
>library('ggplot2')
提示:* Biobase R包具有exprs()、pData()和fData()等函数,可利用它获取基因芯片上的表达数据、样品处理分组等信息以及芯片平台的设计注释信息;
* limma包用于差异相关分析。
* ggplot2用于绘制火山图。
4. 手动下载GEO matrixfile载并初始化数据(示例性数据GSE70213)
首先打开GEO站点(https://www.ncbi.nlm.nih.gov/geo/),在搜索框中输入GSE70213,点击搜索,可获得GSE70213的记录页面。此时,找到页面底下的“Series Matrix File(s)”,点击下载,将数据压缩包保存到事先设置好的R用户目录下(如我设置的为C:\Users\liumwei\Documents\R),然后解压可获得GSE70213_series_matrix.txt文件,如下图:
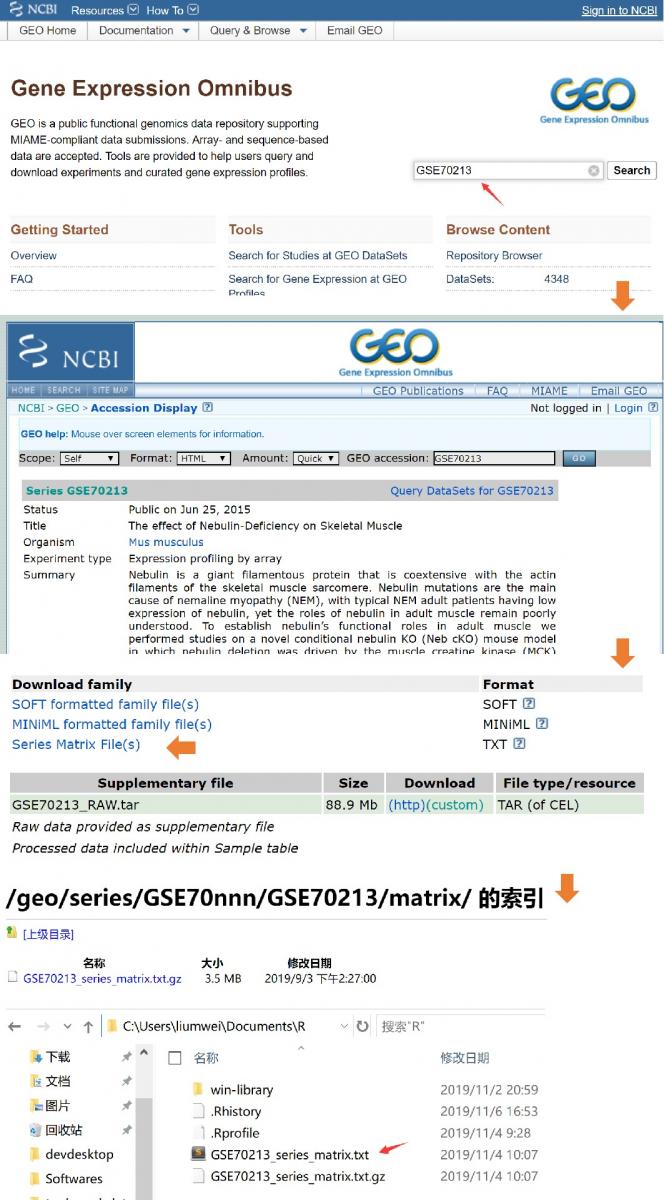
同时,在数据下载时,应对该数据的实验设计等信息充分了解(了解实验目的和设计很重要!),如下图:

可见,该数据集来自小鼠中的一种丝状蛋白Nebulin(与骨骼肌肌动蛋白丝共存的一种蛋白)研究。 尽管目前,先知Nebulin突变是导致Nemaline肌病(NEM)的主要原因,并在典型NEM成年患者中Nebulin低表达。不过,Nebulin在成年肌肉中的作用仍然知之甚少。 故,该研究目的是确定成年肌肉神经元蛋白的功能,模型上采用一种新型的条件性神经元蛋白KO(Neb cKO)雄性小鼠,实验设计上采用两组对照-疾病组设计,即quad-cKO vs quad-control 以及soleus-cKO vs soleus-control,且每个组分别6个重复。
在后面的表型数据分组时,cKo与control可作为表型标签进行提取。
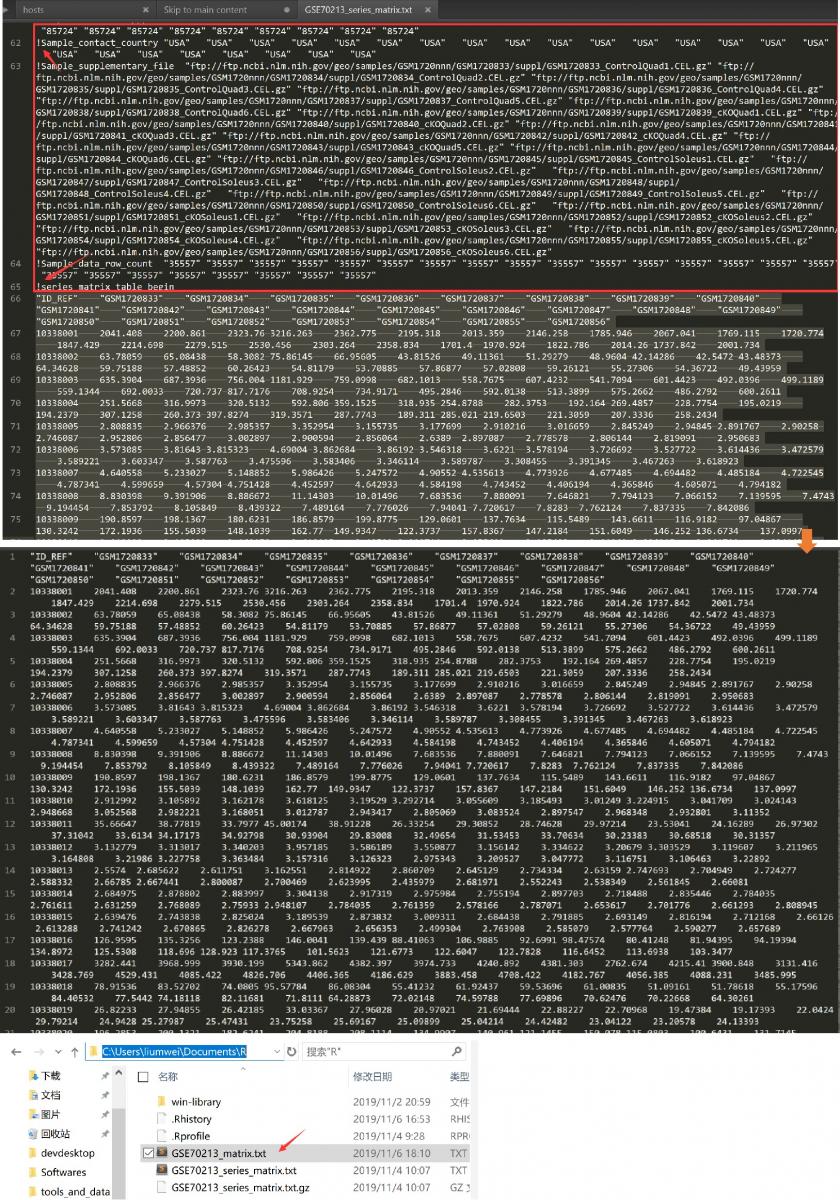
5. 表达值矩阵获取生成
采用Excle、Notepad+++这样的工具打开GSE70213_series_matrix.txt,然后将文件中的首位中含有!的行全部删除,只留下从"ID_REF"开始的数据行(如下图),然后另存为GSE70213_matirx.
 txt
txt
将GSE70213_matirx.txt用Excel打开,数据形式如下:

然后另存为GSE70213_matrix.csv(也就是保存为csv格式。该操作务必进行!如果打开excle打开该文件,发现数据集中在一列里,还需进行数据分列操作。)。
6. 将上面生成的csv数据读入(RStudio Console中操作),然后进行log2转换
>GSE70213_expre <-read.csv(GSE70213_matrix.csv', ,header=TRUE,row.names=1) #或者采用下面的命令读入亦可
>show(GSE70213_expre) #可以查看读入的数据结果究竟是怎么样的。后面的每个命令操作过程,均可用这个命令进行查验。这也是初学者必备的素质。
>GSE70213_expre<-read.table('GSE70213_matrix.csv',header=TRUE,row.names=1) #需要注意
>GSE70213_log2 <- log2(GSE70213_expre) #将表达值进行对数转换
提示:对表达矩阵值进行对数转换,是否需要,其实可以先肉眼观察一下数据。一般数值中出现较大数据和过小数据,最好先进行转换。当然,来自GEO2R有一个自动判断脚本,经修改后如下:
qx <- as.numeric(quantile(GSE70213_expre, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||
(qx[6]-qx[1] > 50 && qx[2] > 0) ||
(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)
if (LogC) { GSE70213_expre[which(GSE70213_expre <= 0)] <- NaN
exprSet <- log2(GSE70213_expre)
print(“log2 transform needed”)}
else{print(“log2 transform not needed”)}
还需要注意的是,如果表达矩阵中出现负数,在进行log转换时,需要提前处理:要么舍弃,要么加上一个固定值(这个一般有商业公司的芯片数据处理手册提供指南)转化成正数。或者取负数的绝对值进行log转换,然后再把负号加上。相关的代码如下:
GSE70213_matrix_abs <- function(x){
x[x==0] <-1
si <- sign(x)
si * log2(si*x)}
GSE70213_expres_abs<-GSE70213_matrix_abs(GSE70213_expre)
7. 基于箱线图进行数据整体情况调查
>boxplot(data.frame(GSE70213_log2),col="red")
结果如下:
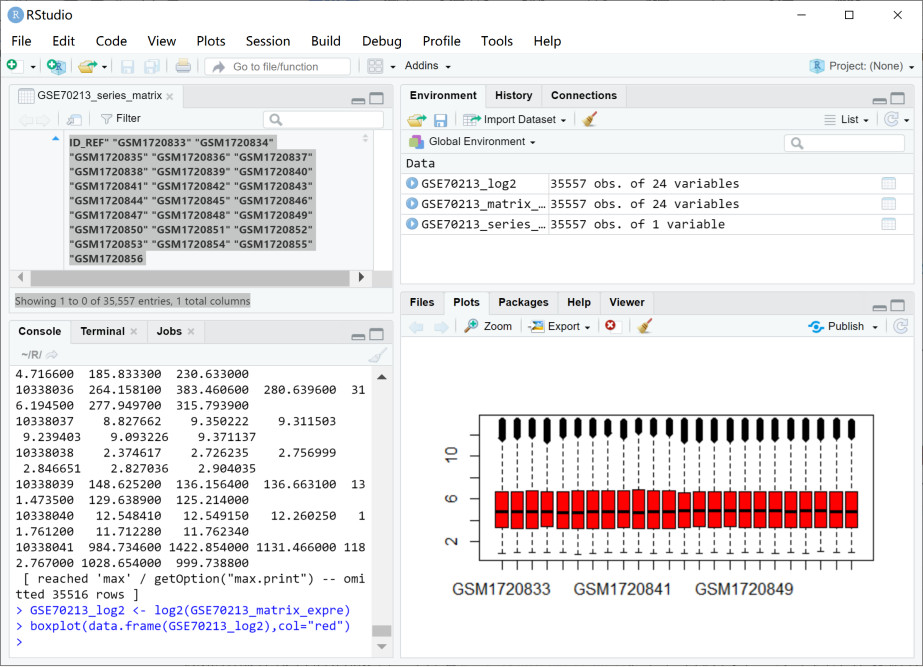
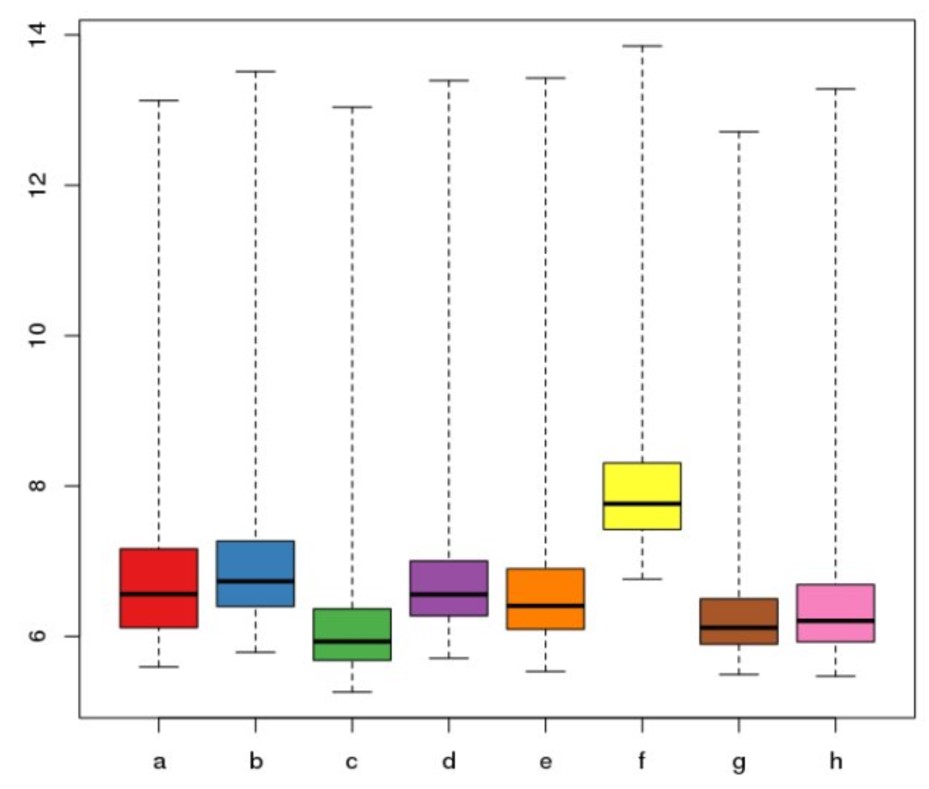
从箱线图可以看出,样本间的基因表达数据一致性还是很高。比如中位值均在5附近,最高值和最低值基本接近。不过,其他有些数据整齐可能很不好,甚至会出现下图的结果。尽管如此,我们看到数据整齐性还可以在优化一下。优化的最好办法是对数据进行标准化操作(至于为何要标准化操作及背后的原理,可查看这个页面的解释说明。),再进入下一步基因差异数据分析:
标准化是组学数据最基本的问题,有多种方法可以进行。在此,我们采用limma包中normalizeBetweenArrays函数来重新标准化,相关命令和结果如下:
GSE70213_expr_N <- normalizeBetweenArrays(GSE70213_expre)
GSE70213_N_log2 <- log2(GSE70213_expr_N)
boxplot(data.frame(GSE70213_N_log2),col="red")
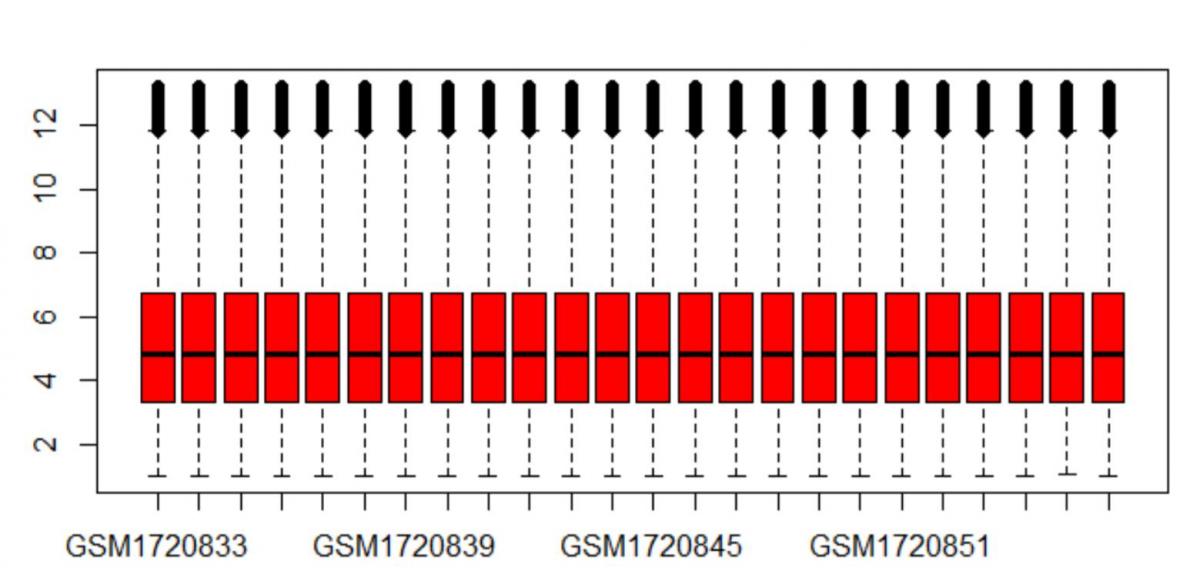
可见,重新标准化的数据,其整齐度更好。
8. 基因差异分析
A. 数据分组准备
根据GSE70213的页面样本信息(表型信息)对数据进行手动分组准备(下图),保存为csv格式(GSE70213_type.csv)
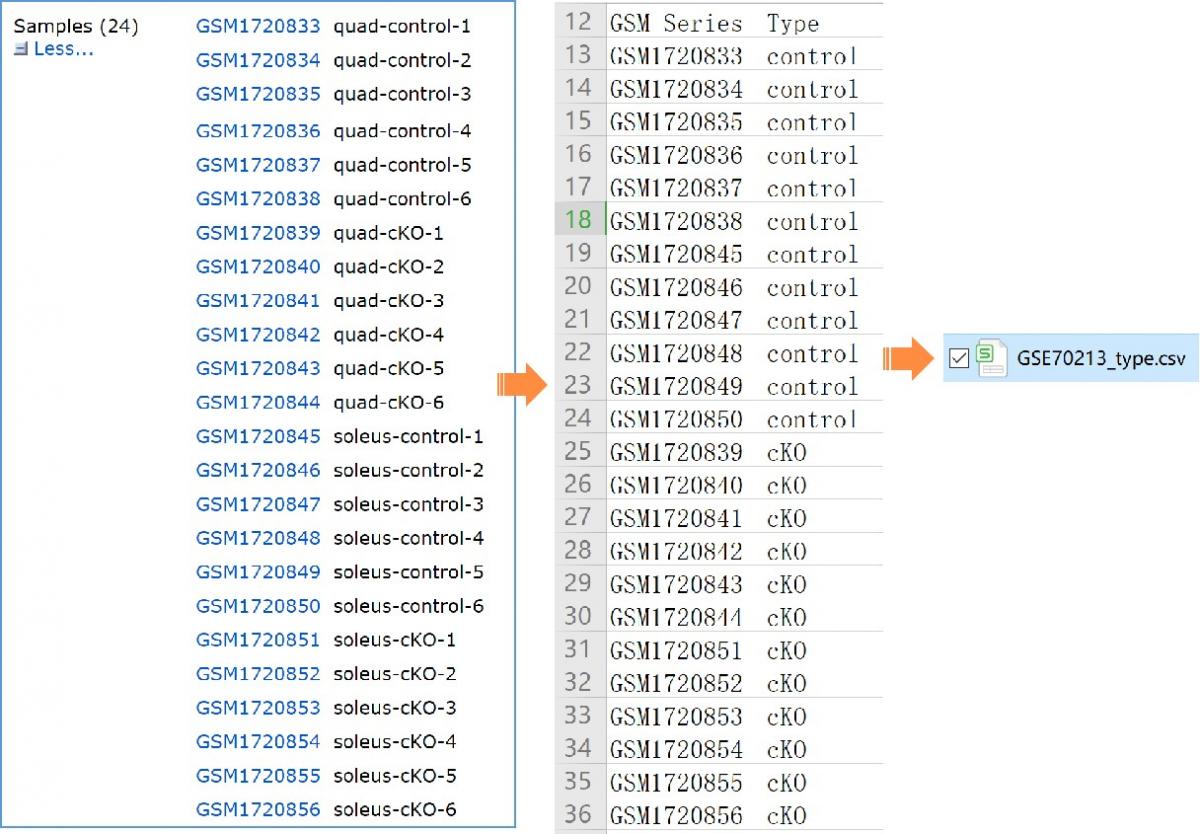
准备好上面的表型分组数据后,在RStudio中将其载入到内存中:
>GSE70213_phenodata <- read.table('GSE70213_type.csv', header=TRUE, row.names=1, check.names = FALSE)
提示:* 采用read.table()将表型分类数据表读入内存中。 header=TRUE表示要检查表头;row.names=1表示把第一列设为行名。
* 注意,如果出现如下错误,这表明在"row.names=1"前输入了不能识别的符号(比如中文的空格)。为避免这个问题,请确认输入命令时,请切记将输入法切换到英文状态,并使用删除键,试着将"row.names=1"可能的空格删除。这是初学者常常会出现的错误和问题。在前面的命令输入时,也特别注意这个问题。
错误: unexpected input in "GSE70213_phenodata <- read.csv('GSE70213_type.csv', header=TRUE,\"
B. 构建分组矩阵
>GSE70213_group <- GSE70213_phenodata [,1] #获取第二列的表型标签
>GSE70213_design <- model.matrix(~0+factor(GSE70213_group)) #将表型标签进行数字0和1的转换
>head(GSE70213_design) #查看表型标签进行数字转换后,整个表型表变换的结果,它和show()的结果是异曲同工的
>colnames(GSE70213_design) <- levels(factor(GSE70213_group)) #将表型标签作为列
>rownames(GSE70213_design) <- colnames(GSE70213_N_log2) #将对数表达值作为行
提示:colnames和rownames分别表示设置分组举证的列和行
C. 基因差异矩阵数据
>GSE70213_cont.matrix <- makeContrasts(paste0(unique(GSE70213_group),collapse="-"),levels=GSE70213_design)
提示:makeContrasts()构建比较矩阵
D. 差异分析
>GSE70213_fit <- lmFit(GSE70213_N_log2,GSE70213_design) #给定系列数据,为每个基因进行线性模型拟合)
>GSE70213_fit2=contrasts.fit(GSE70213_fit,GSE70213_cont.matrix) #基于拟合线性模型进行系数估计和标准误差计算
>GSE70213_fit3 <- eBayes(GSE70213_fit2)#基于经验贝叶斯进行进行t统计量、F统计量和差异对数表达计算
>GSE70213_output = topTable(GSE70213_fit3,coef=1,n=Inf,adjust="BH") #从线性模型拟合结果中,进行排序,并提取排序靠前的基因列表
>GSE70213_nrDEG = na.omit(GSE70213_output) #删除基因排序列表中出现的缺失值,并将结果返回
提示:R中通过na.fail和na.omit来处理样本中的缺失值。na.fail()用于先判断,后返回结果:如果样本中有缺失值,回返回错误,如果没有,则返回结果。而na.omit()则表示将缺失值删除后,把结果返回。attr(na.omit(),"na.action")表示对缺失值进行检查,并返回缺失值的下标;is.na():判断向量内的元素是否有缺失(NA)
F. 差异分析结果文件
>write.table (GSE70213_nrDEG, paste("GSE70213_DE.csv")) #用下面的命令,也能将结果存到文档中
>write.table(GSE70213_nrDEG, "GSE70213_DE.csv", quote=F, sep=",") #该命令加入了相关文档数字分隔的字符定义
提示:csv 文件一般是用逗号分隔的,故而 sep = ",";tsv 文件是用制表符分隔的,故而 sep = "\t";常用的分隔符还有空格 sep = " "
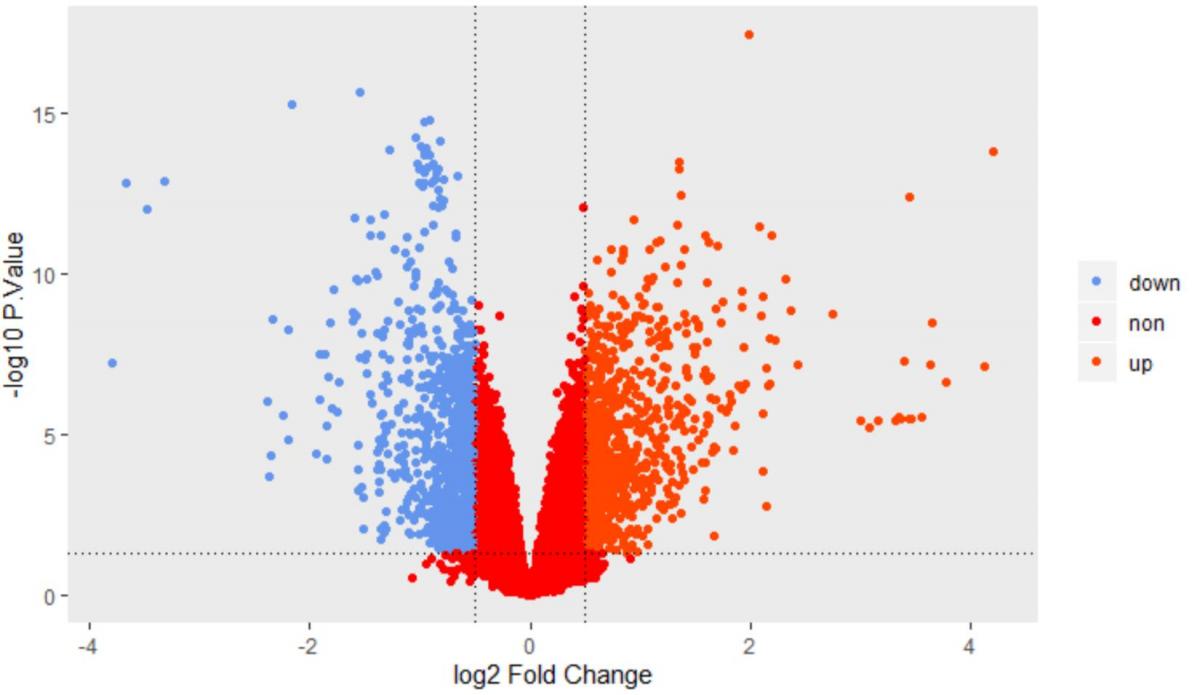
9. 差异基因火山图可视化结果
>GSE70213_alldiff<-read.csv("GSE70213_nrDEG.csv",header=TRUE,row.names=1,check.names=FALSE)
>GSE70213_alldiff$threshold[GSE70213_alldiff$P.Value < 0.05 & GSE70213_alldiff$logFC > 0.5] = "up"
>GSE70213_alldiff$threshold[GSE70213_alldiff$P.Value < 0.05 & GSE70213_alldiff$logFC < -0.5] = "down"
>GSE70213_alldiff$threshold[GSE70213_alldiff$P.Value > 0.05 | (GSE70213_alldiff$logFC >= -0.5 & GSE70213_alldiff$logFC <= 0.5)] = "non"
>ggplot(GSE70213_alldiff,aes(x=logFC,y=-log10(P.Value),colour=threshold))+ xlab("log2 Fold Change")+ ylab("-log10 P.Value")+ geom_point()+ scale_color_manual(values =c("#6495ED","red","#FF4500"))+ geom_hline(yintercept=1.30103,linetype=3)+ geom_vline(xintercept=c(-0.5,0.5),linetype=3)+ theme(legend.title = element_blank(),panel.grid.major = element_blank(),panel.grid.minor = element_blank())
提示:GSE70213_alldiff$threshold用于设置取值“up”、“down”和 “non”;ggplot()绘图函数,其中P.Value和logFC的值需根据研究具体情况而定。这里,分别被设置为0.05和0.5,aes()为坐标轴取值设定,xlab和ylab为横纵坐标名称设定,scsle_color_manual为设定颜色值,geom_hline和vline为分割线设定,theme为主题设定。